Sistemas polimórficos de interesse antropológico, médico e forense
| Site: | UFPR Virtual - o Moodle oficial |
| Curso: | BG048 GENÉTICA DE POPULAÇÕES HUMANAS - BIOMED |
| Livro: | Sistemas polimórficos de interesse antropológico, médico e forense |
| Impresso por: | Usuário visitante |
| Data: | Saturday, 8 Aug 2026, 18:25 |
Descrição
Conteúdo do módulo
1. Definição de polimorfismo
O genoma humano possui cerca de 3 bilhões de pares de bases (pb), embora essa quantidade varia de um indivíduo para outro. Entre dois genomas haploides existe cerca de 0,1% de variação que pode ser classificada em tipos, que veremos à continuação. O conjunto de alelos (formas alternativas) de uma sequência de DNA em uma população constitui o pool gênico da mesma. Este funciona como um reservatório de diversidade genética que pode servir como material para adaptação a mudanças no ambiente, por exemplo.
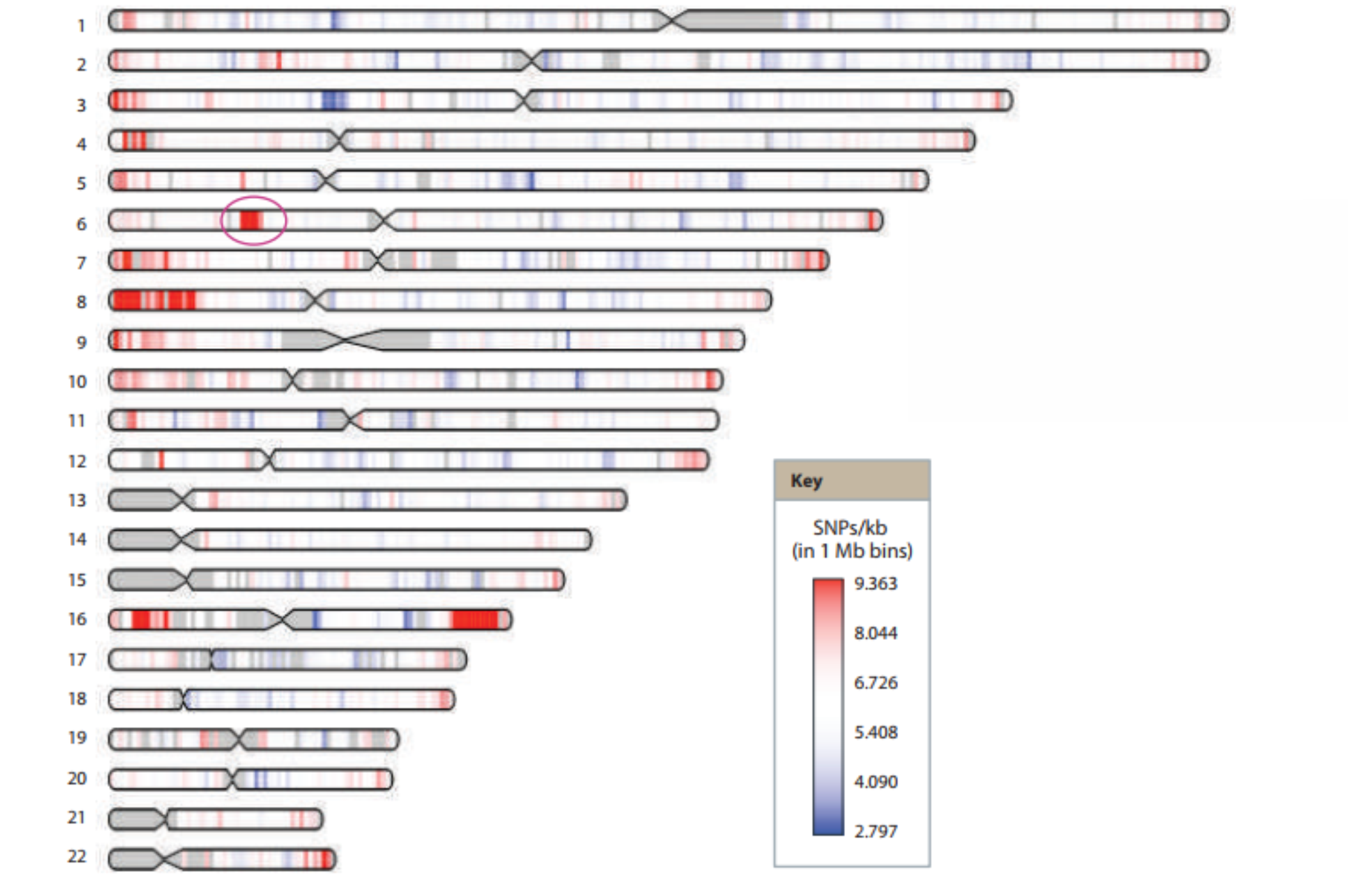
Dizemos que uma sequência é polimórfica quando a mesma apresenta, no mínimo, dois alelos na população, cada um com frequência >= 1%. Sequências polimórficas podem ser bialélicas (Ex.: inserção/deleção, substituições com dois alelos) ou multialélicas (Ex.: polimorfismos de número de cópias). O contrário de polimorfismo é o monomorfismo (ou sequência monoalélica) onde não ocorre variação. O conceito de polimorfismo é qualitativo, podendo uma sequência ser ou não ser polimórfica. Assim, o que pode variar entre distintas sequências é o seu grau de polimorfismo. Exemplo: como se observa na figura o loco HLA (do inglês Human Leucocite Antigen, antígeno leucocitário humano) destacado com um círculo vermelho, representa uma das regiões com maior densidade de polimorfismos de nucleotídeo único, ou SNPs. Dizemos que este loco possui um alto grau de polimorfismo.
É importante diferenciar o conceito de variante. O termo variante pode ser utilizado para falar de um polimorfismo, mas o primeiro também abrange diferenças menos frequentes entre cópias do genoma cujos alelos não necessariamente atingem a frequência necessária para ser incluído na categoria de polimorfismo. Ex.: variantes podem ser singletons (um dos alelos foi detectado apenas uma vez na população) ou doubletons.
Por último o termo mutação indica uma mudança de novo (que ocorre pela primeira vez). No momento que ela ocorre existe como singleton. O resultado dos processos evolutivos fará com que essa mutação siga um dentre vários caminhos, desde aumentar a sua frequência e se tornar um polimorfismo até ser eliminada da população.
O que é necessário para que uma mutação se torne um polimorfismo?
Mutações são o resultado de danos ao DNA e falhas no sistema de reparo. A diversidade em um dado locus (ou local no genoma, em plural loci, em português loco) depende tanto da sua taxa de mutação como da seleção natural e da história populacional. Para que uma mutação se torne um polimorfismo esta deve:
- Ocorrer na linhagem germinativa (células que dão origem aos gametos).
- Permitir a sobrevivência e não afetar a reprodução. Ex.: Mudanças no número de cromossomos (aneuploidias) decorrem de falhas na segregação cromossômica na meiose por isso não são herdáveis.
- Cada alelo deve alcançar uma frequência >= 1% na população.
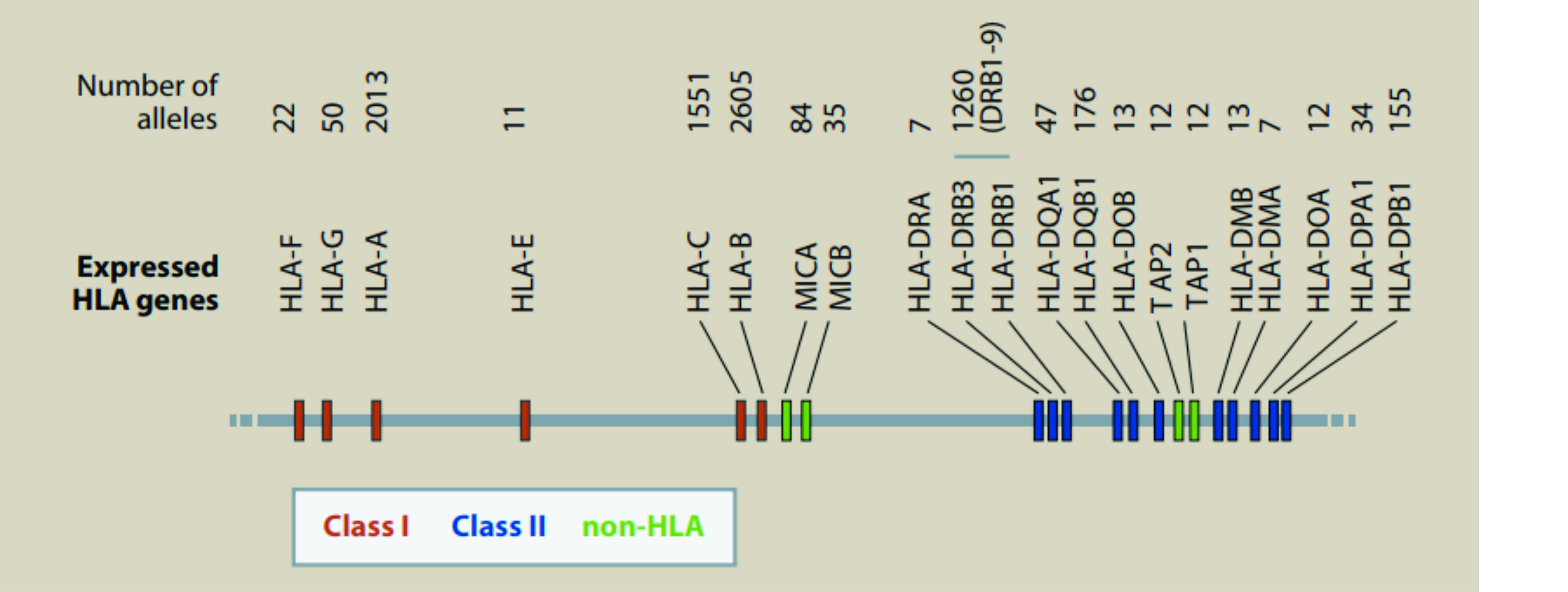
Os genes HLA apresentam antígenos aos linfócitos T, estando envolvidos com a resposta imune adaptativa. Diferentes alelos de cada gene individual variam na habilidade de apresentar antígenos de diferentes patógenos. O locus HLA (3,6 Mb no cromossomo 6) está dividido em 3 regiões, chamadas "classes". Nas regiões de classe I e II, antigos eventos de duplicação geraram distintos genes expressos e pseudogenes.
Os diferentes alelos são mantidos na população por seleção balanceadora, permitindo que a mesma esteja preparada para a sobrevivência a patógenos emergentes. Hoje são os principais determinantes da histocompatibilidade.
Tipos de marcadores genéticos
A começos do século XX, após a redescoberta dos trabalhos de Gregor Mendel (1822-1884) sobre hibridização de plantas, começa um período de extensa pesquisa no campo da genética de populações. Porém, durante essa primeira etapa os pesquisadores ainda não tinham as ferramentas para estudar diretamente os polimorfismos genéticos em humanos. Os primeiros trabalhos da área são voltados à observação indireta da variação genética, isto é, no nível das proteínas (produtos do DNA). Esse primeiro grupo de marcadores estudados são hoje chamados de Marcadores Clássicos e foram a base das primeiras descobertas em poliformismos.
Marcadores Clássicos
No grupo de marcadores clássicos podemos diferenciar dois subgrupos em função da forma de estudo dos mesmos:
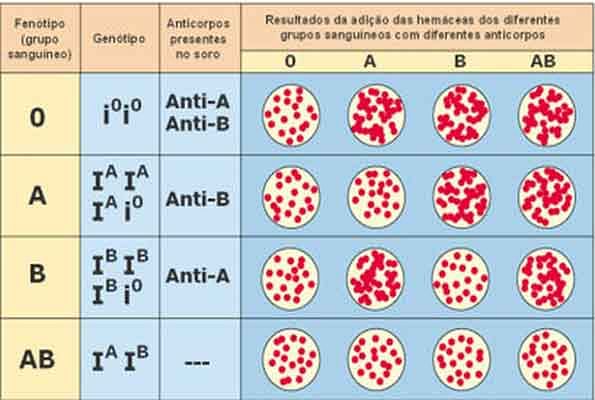
Marcadores imunogenéticos: a detecção da variação é baseada no reconhecimento de um antígeno por um anticorpo. Neste caso é o antígeno quem indica a variação no nível do DNA. O estudo pioneiro neste campo foi o realizado pelo casal polonês Hanka e Ludwik Hirszfeld, na Sérbia, durante a Primeira Guerra Mundial. Eles estudaram os grupos sanguíneos ABO em 8000 pessoas de diferentes nacionalidades. Outros estudos relevantes da época foram voltados para alelos HLA e para imunoglobulinas.
Marcadores bioquímicos: a variação genética, refletida em mudanças na cadeia de aminoácidos, é detectada através do padrão de migração da proteína em um gel de eletroforese. Isto se deve a que mudanças na cadeia de aminoácidos afetam a carga elétrica e a conformação da proteína, o que afeta a sua velocidade de migração no gel. Porém, essa abordagem apenas é útil quando a proteína a ser estudada se encontra em alta concentração, permitindo a sua visualização direta no gel, o que não é o caso de muitas proteínas humanas. Essa limitação foi contornada pelo uso de outro sistema de detecção, a tinção histoquímica. Neste caso, a proteína estudada deve ser uma enzima e sua presença no gel é detectada por um produto de reação de fácil visualização, como compostos coloreados. Desta forma, as mudanças no padrão de migração de proteínas em baixa concentração podem ser visualizadas.
Marcadores de DNA
A detecção direta de variações na sequência de nucleótidos do DNA começa a ser possível graças à descoberta, em 1970, das enzimas de restrição. A possibilidade de cortar e colar fragmentos de DNA da origem aos primeiros marcadores de DNA estudados: os polimorfismos de comprimento de fragmentos de restrição (ou RFLP, do inglês Restriction Fragment Lenght Polymorphism). Estes marcadores baseiam-se no reconhecimento pela enzima de restrição de sequências específicas de nucleótidos e na produção, após o corte, de fragmentos com comprimentos diferentes. A visualização dos fragmentos é realizada através da técnica de Southern blotting: o produto de reação enzimática é corrido em um gel de agarose e posteriormente transferido, por capilaridade, a uma membrana de nitrocelulose. Essa membrana é incubada com sondas de DNA marcado com nucleótidos radioativos, os quais são detectados pela exposição a raios-X. Fragmentos de DNA de comprimento conhecido, e também marcados radioativamente, são incluídos na corrida eletroforética inicial, permitindo estimar o tamanho dos fragmentos de restrição.
2. Tipos de polimorfismo genético
Existem várias categorias de polimorfismo genético, cada uma das quais possui mecanismos de origem e aplicações específicas que dependem das suas características. Podemos diferenciar:
Polimorfismo de nucleotídeo único: ou SNPs, do inglês Single Nucleotide Polymorphisms. Nessa categoria incluímos substituições ou indels (inserção/deleção) de uma única base.
Polimorfismo de número variável de repetições em tandem: ou VNTRs, do inglês Variable Numer of Tandem Repeats. Essa categoria abrange sequências que mudam no número de cópias de um fragmento repetitivo. Por sua vez, as VNTR podem ser classificadas em: satélites (a sequência repetida possui >= 100 pb), minissatélites (8-100 pb) e microssatélites (a sequência repetida possui 1-7 bp).
Elementos transponíveis: também chamados elementos moveis ou elementos repetitivos dispersos. São sequências que inserem cópias de se mesmas em outras partes do genoma. Elas compõem 45% do genoma humano.
Variantes estruturais: este grupo inclui inversões e translocações balanceadas ou não balanceadas (como indels) que abrangem fragmentos maiores a 1kb.
Das categorias mencionadas, SNPs, microssatélites e algumas classes específicas de elementos transponíveis são amplamente utilizadas na genética de populações devido a sua maior facilidade de estudo. Uma característica importante dos distintos polimorfismos que afeta enormemente a sua aplicabilidade nas distintas áreas da genética é a sua velocidade de mutação. A velocidade de mutação de um polimorfismo permitirá que ele seja utilizado, ou não, para identificar variação no nível inter-individual (diferenças entre os indivíduos e ainda entre parentes cercanos) ou no nível populacional.
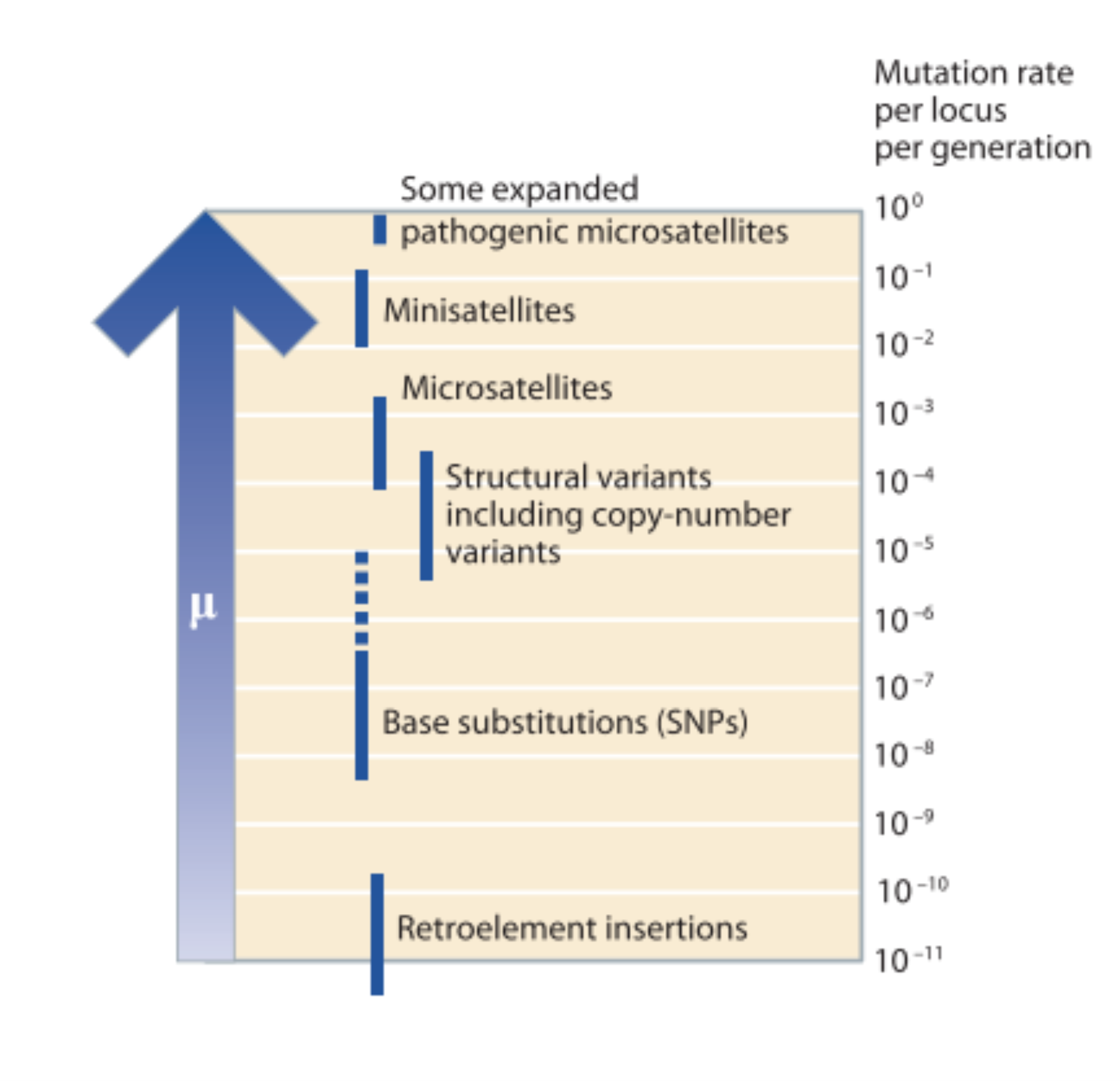
Mecanismos de origem e características dos polimorfismos genéticos
SNPs
Os SNPs representam a forma mais simples de polimorfismo genético, afetando uma única base do DNA. Embora nessa categoria estejam incluídas indels de úma única base, o termo SNP é mais comumente utilizado para falar de substituições de bases. Os estudos realizados até o momento em populações humanas mostraram que existem, em média, ~3-4 milhões de SNPs por genoma. Estudos em populações europeias mostraram ser esperado encontrar um SNP a cada 1250 pb. No caso de genomas africanos, essa frequência aumenta a um SNP a cada 1006 pb, um claro indicador da maior diversidade genética africana quando comparada à diversidade encontrada nos demais continentes.
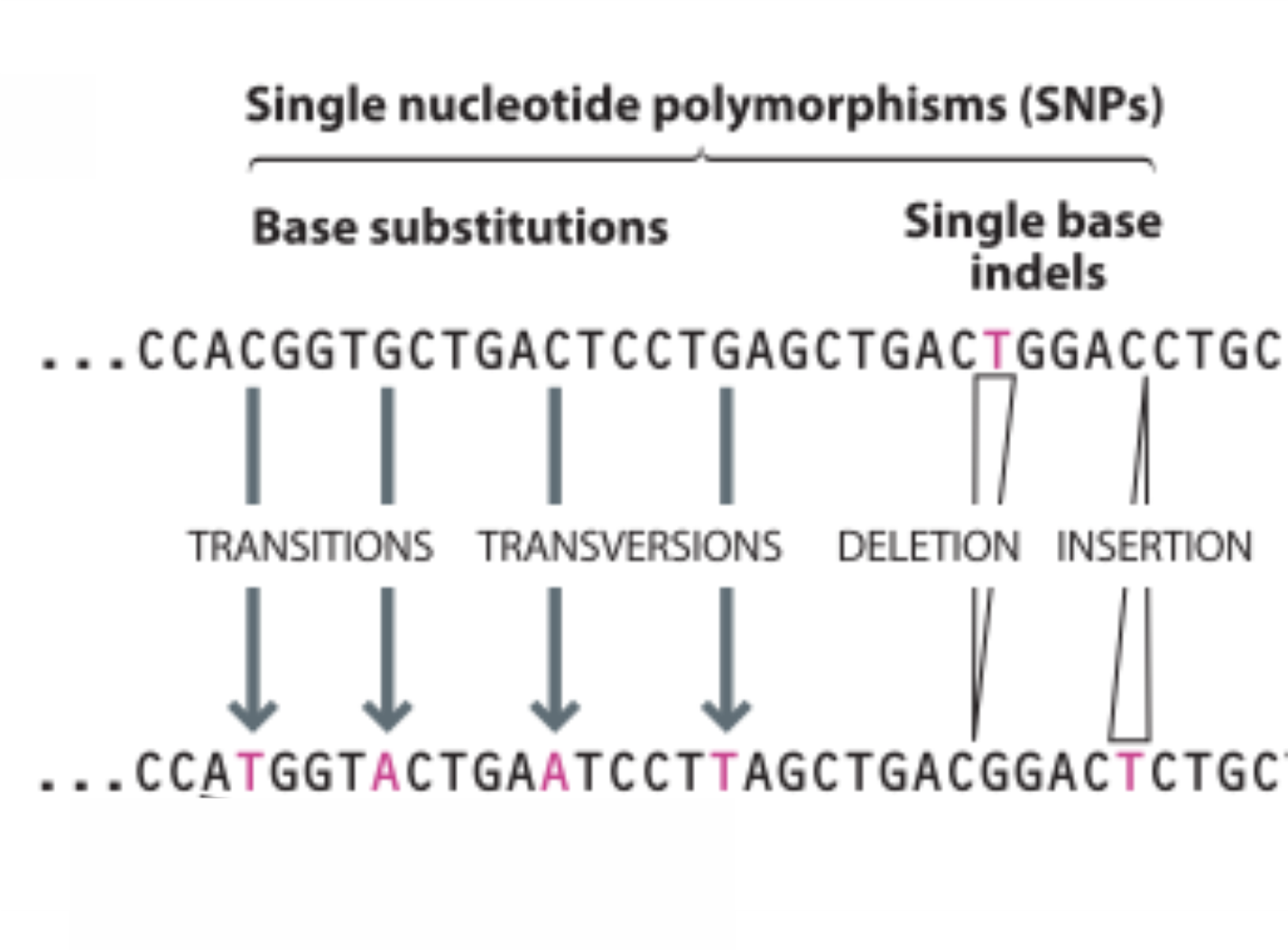
Nomenclatura
Os SNPs são identificados por um número rs, do inglês Reference SNP. Ex.: rs334 (o SNP que determina a anemia falciforme).
Características
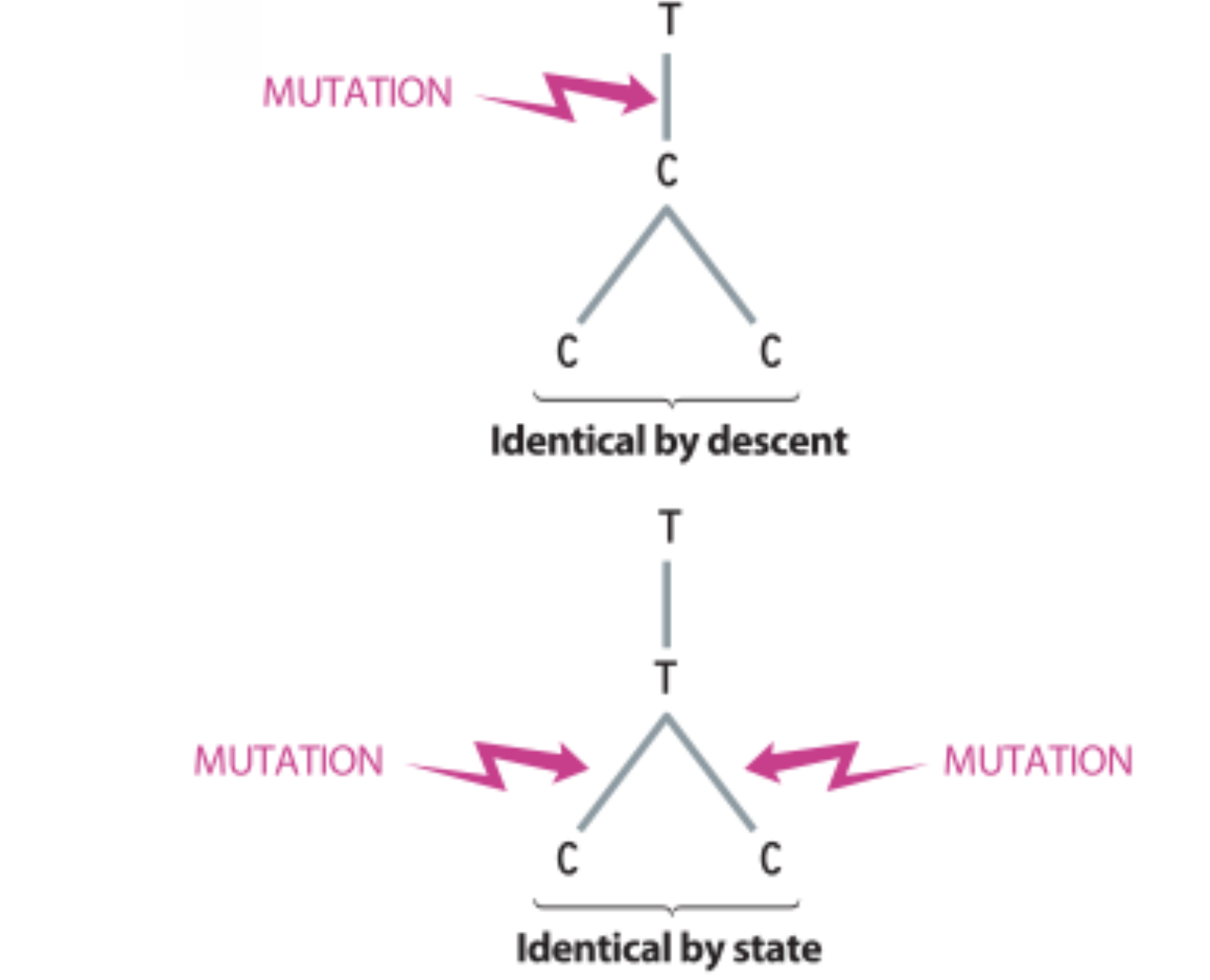
Os SNPs ocorrem em todos os cromossomos (autossomos, Y, X, DNAmt). Possuem uma baixa velocidade de mutação, motivo pelo qual, embora em teoria possam ocorrer 4 alelos por cada locus, os SNPs são comumente bialélicos. Os casos de SNPs trialélicos são raros, e, se ocorrem, o terceiro alelo é encontrado em frequências muito baixas e em populações específicas. Também em decorrência da sua baixa velocidade de mutação, os SNPs apresentam identidade por descendência (IBD, do inglês identity by descent). Isto significa que a existência do mesmo alelo de um SNP em dois indivíduos decorre necessariamente de ambos terem sido herdados de um ancestral comum. O alelo ancestral pode ser deduzido olhando o alelo existente nos grandes macacos. O alelo alternativo se denomina alelo derivado. Os SNPs são comumente descritos pelo MAF, ou Minor Allele Frequency, ou a frequência do alelo menos comum. Quando ambos os alelos ocorrem com uma frequência >= 5% se denominam variantes comuns.
O mecanismo de origem do SNP é uma mutação, que pode ser decorrente da ação de agentes mutagênicos exógenos, como agentes físicos (Ex.: radiações de alta penetrância nos tecidos como raios-X ou raios gamma) ou químicos (Ex.: brometo de etídeo, mitomicina C, etc). É importante destacar que, para que qualquer agente mutagênico externo possa dar origem a um SNP ele deve atingir a linha germinal, portanto deve penetrar os tecidos e chegar até as gonadas. Por outro lado, mutações podem decorrer de mecanismos endógenos, como erros durante a replicação do DNA. A ocorrência da mutação por si só não garante que a mesma será mantida, portanto, é necessário também que ocorram falhas no sistema de reparo.
Métodos de estudo de SNPs
Dentre os métodos utilizados atualmente para o estudo de SNPs podemos diferenciar entre aqueles que são específicos para este tipo de polimorfismo, como PCR-RFLP, PCR-SSP e microarranjo de DNA, e aqueles que são aplicáveis ao estudo de qualquer tipo de polimorfismo genético e que se baseiam no sequenciamento do DNA. Entre os métodos específicos podemos mencionar (embora não se limitam aos mencionados aqui):
PCR-RFLP: este método consiste em uma atualização do RFLP antes descrito, ao qual é incorporada uma etapa inicial de amplificação por PCR (do inglês Polymerase Chain Reaction) do fragmento de interesse, onde se encontra o sítio polimórfico. Após a amplificação, o produto de PCR é incubado com enzimas de restrição que reconhecem e cortam a sequência ao redor do SNP. A presença de um dos alelos evita o reconhecimento e o corte pela enzima, gerando um fragmento maior e reconhecível pelo seu tamanho em um gel de eletroforese. O gel pode ser então corado com corantes específicos de DNA. A prévia amplificação do fragmento permite que haja uma quantidade suficiente de DNA para que as bandas possam ser visualizadas em um transiluminador.
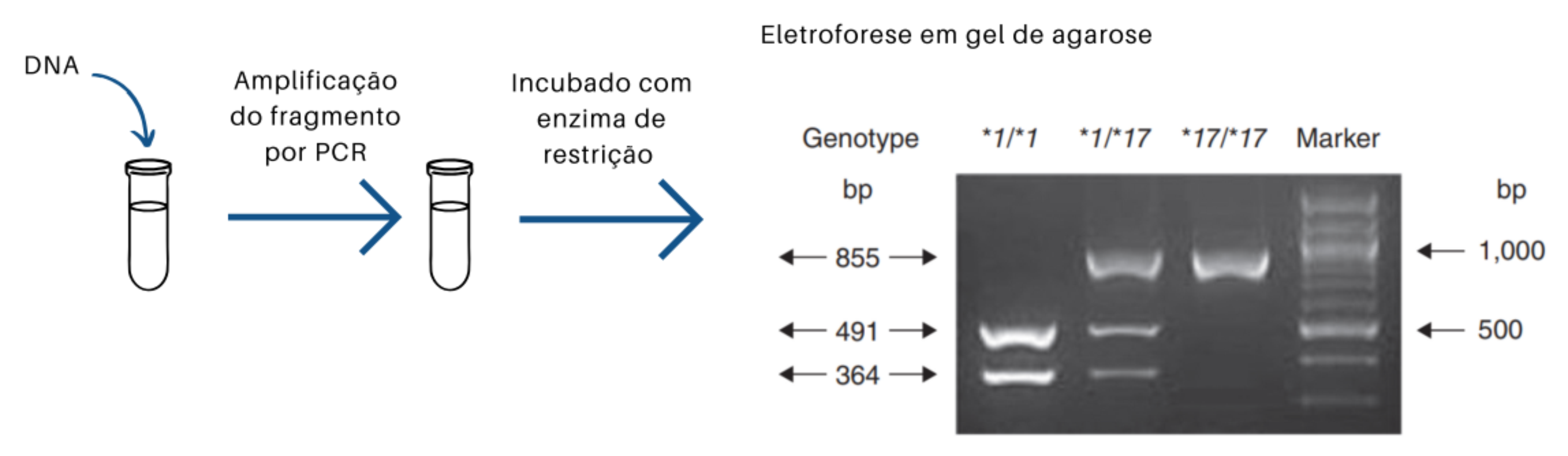
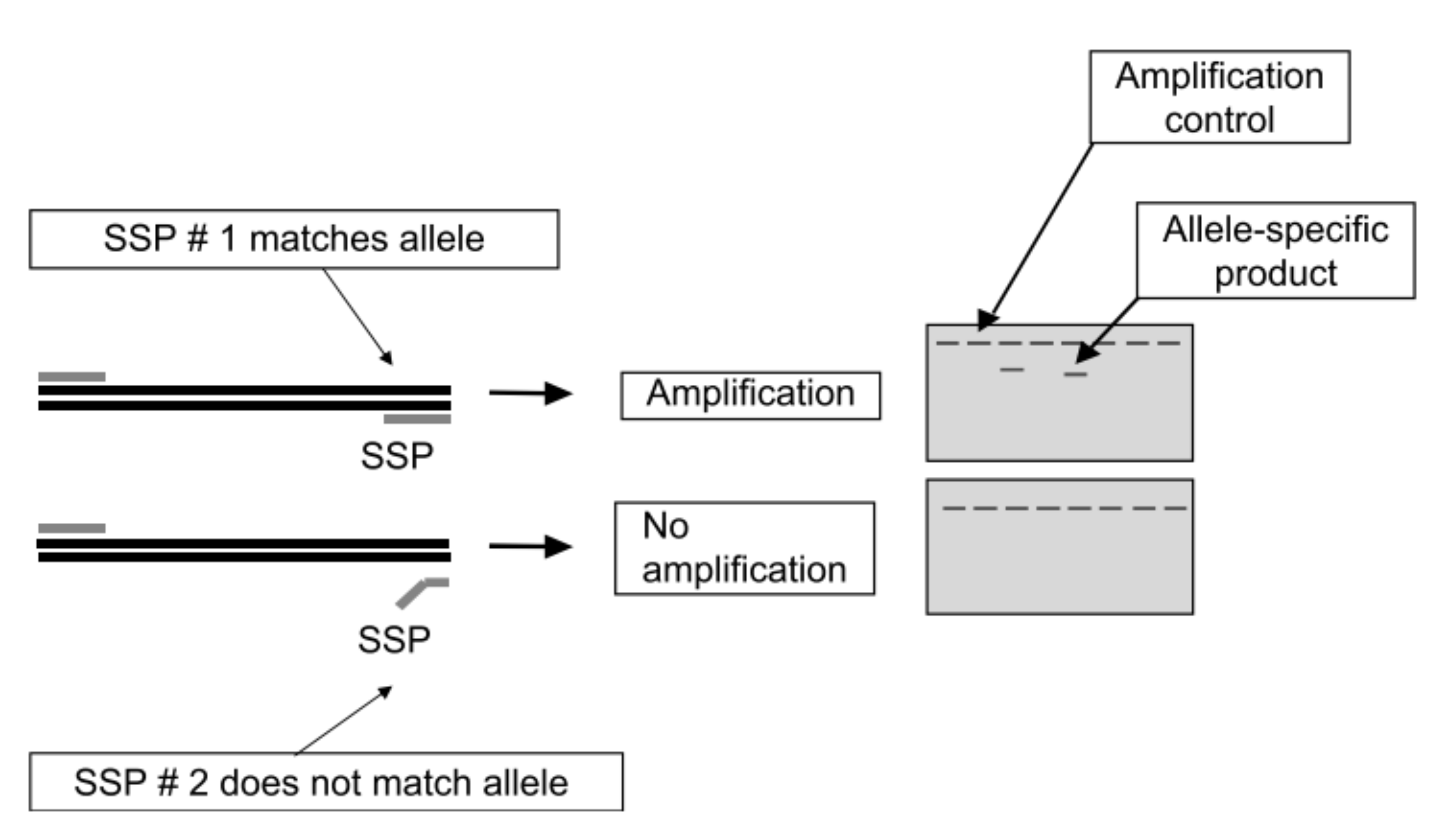
PCR-SSP: ou PCR com iniciadores específicos de sequência (do inglês, Sequence Specific Primer). Esta técnica utiliza iniciadores específicos para cada um dos alelos de um SNP em uma mesma reação de PCR. Desenham-se iniciadores cujos extremos 3 ́ devem hibridizar cada um com apenas um dos alelos do SNP, e um terceiro iniciador comum para ambas as reações. Após a reação os tamanhos dos fragmentos se visualizam por eletroforese. Assim, indivíduos homozigotos terão amplificação a partir de apenas um dos iniciadores, enquanto indivíduos heterozigotos terão amplificação a partir de ambos os iniciadores, observándo-se duas bandas no gel.
Microarranjos de DNA
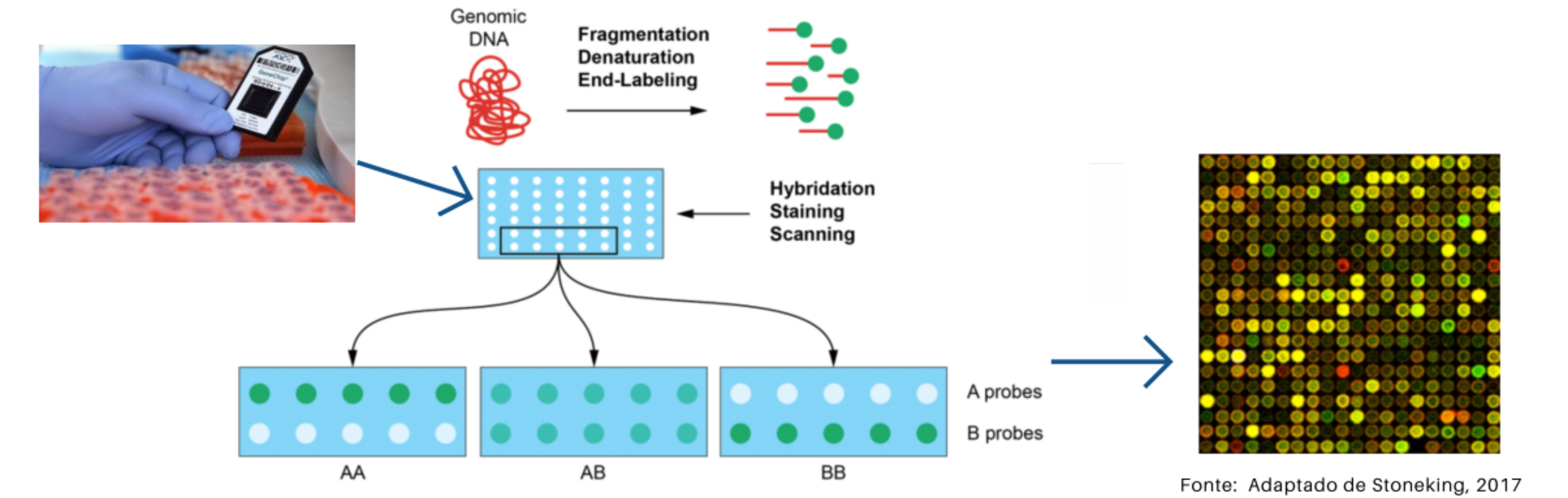
Também chamados "chips". Os microarranjos permitem a genotipagem de milhares de SNPs para cada amostra, geralmente abrangendo "Tag" SNPs de todo o genoma (Genome-Wide). O termo Tag SNP (Tag provem do inglês e significa "etiqueta") faz referência ao uso de certos SNPs como marcadores de uma determinada região genômica, a qual é herdada "em bloco" por apresentar desequilíbrio de ligação. Assim, os SNPs que se encontram próximos são herdados em combinações de alelos ou haplótipos (um alelo do SNP1 sempre é herdado junto o com um certo alelo do SNP2), desta forma o conhecimento do estado alélico do Tag SNP (o SNP presente no microarranjo) pode nos informar dos estados alélicos dos SNPs que se encontram no mesmo haplótipo, mas que não foram genotipados no microarranjo. Os chips são produtos comerciais e desenhados para a genotipagem de conjuntos de SNPs pre-selecionados e, como consequência não permitem a detecção de novos SNPs. Em um chip, cada poço possui sondas marcadas com fluorescentes diferentes para cada alelo (verde ou vermelho). O sinal amarelo indica a hibridização com ambas as sondas (o indivíduo é heterozigoto para esse loco). Cada chip permite a genotipagem de uma amostra, mas eles são comumente vendidos por quantidades (>= 100 chips).
Técnicas de sequenciamento
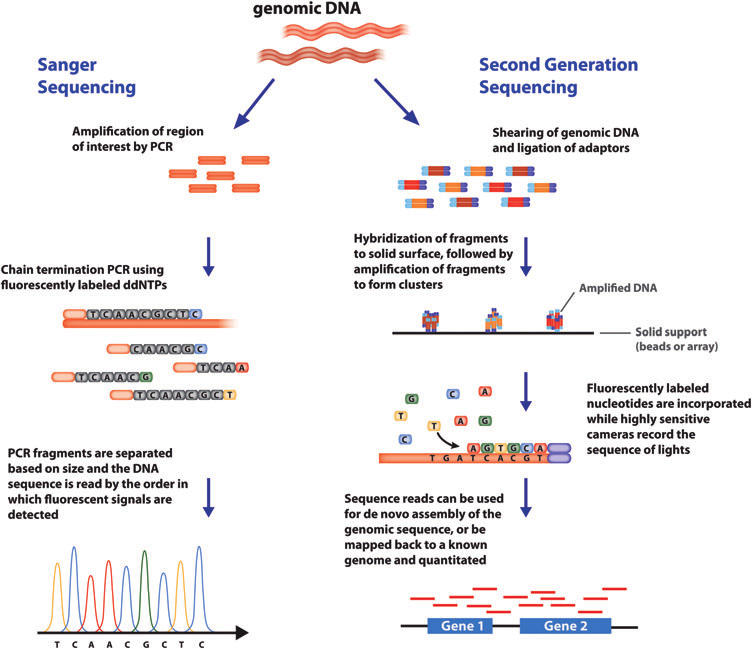
Sequenciamento de Sanger
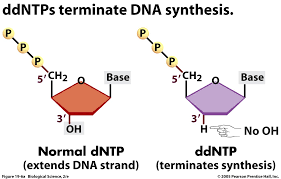
Este método também é conhecido como sequenciamento de primeira geração, método dos dideoxi ou de terminação de cadeia. A técnica permite detectar cada nucleotídeo na medida que este é adicionado
durante a reação de polimerização. Na reação de sequenciamento são utilizados dideoxi nucleotídeos (ddNTPs) marcados com um fluoróforo diferente para cada base (A, T, G ou C), assim como também dNTPs não marcados, estes últimos em maior concentração. Quando um ddNTP é adicionado pela DNA polimerase a reação termina, dado que o ddNTP não possui o 3´-OH necessário para a incorporação de um novo nucleotídeo na cadeia. É gerado assim um fragmento com o comprimento equivalente à posição do último nucleotídeo incorporado e uma cor específica para a última base. Ao final todos os fragmentos são separados em um tubo capilar e detectados pelo sequenciador. O resultado do sequenciamento é lido como um cromatograma onde cada pico representa um nucleotídeo da sequência.
Sequenciamento de nova geração (NGS = Next Generation Sequencing)
Também chamado de sequenciamento de segunda geração. O termo inclui vários métodos que diferem dependendo da empresa que oferece o serviço, já que este requere de um equipamento adequado. As características gerais do NGS incluem uma etapa de fragmentação do DNA genômico (que não precisa ser amplificado previamente como no sequenciamento de Sanger) e uma segunda etapa de imobilização dos fragmentos numa superfície sobre a qual será realizada a reação de sequenciamento. Esses métodos permitem sequenciar genomas completos ou grandes fragmentos e, inclusive, muitas amostras numa mesma reação. Novas abordagens permitem o sequenciamento apenas de regiões exônicas (codificadoras de proteínas).
Microssatélites (ou STR, do inglês Short Tandem Repeats )
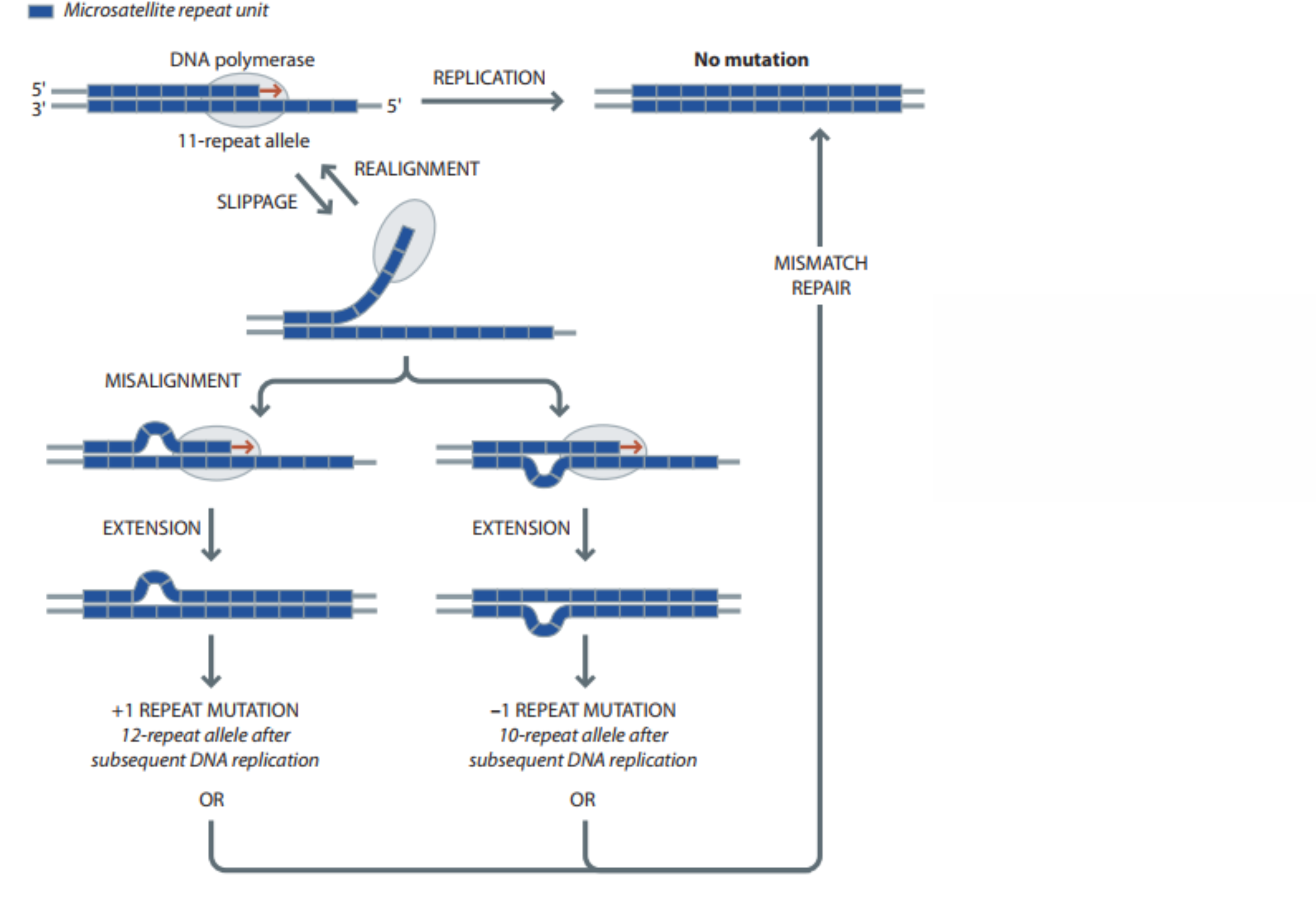
Os microssatélites são repetições de sequências de entre 1–7 bp que, usualmente, se encontram presentes em 10–30 copias. Ocorrem em todos os cromossomos humanos (autossomos, Y, X e mtDNA). Este tipo de polimorfismo possui uma alta velocidade de mutação, motivo pelo qual são comumente multialélicos, sendo os seus alelos os distintos números de cópias existentes. Devido a sua alta velocidade de mutação, alelos com o mesmo tamanho em distintos indivíduos não são necessariamente idênticos por descendência e, sim, idênticos por estado (ou seja, os mesmos alelos em distintos indivíduos originam-se por evolução convergente). Combinações de STRs podem produzir perfis genéticos únicos para um indivíduo, permitindo a sua identificação. Devido à ocorrência de evolução convergente nesses loci o alelo ancestral não pode ser deduzido olhando o alelo existente nos grandes macacos (pois o número de cópias pode ter-se reduzido o aumentando múltiplas vezes durante a evolução).
O mecanismo de origem dos STR são erros no alinhamento das fitas de DNA durante a replicação. Devido a sua natureza repetitiva, sequências microssatélites podem hibridizar consigo mesmas, gerando forquilhas e encurtando a fita. Se a fita onde se forma a forquilha é a mesma fita que está sendo polimerisada então um número de repetições proporcional ao número que compus a forquilha será adicionado. Se a fita onde a forquilha é formada é a fita molde, a cadeia replicada terá menos repetições que a cadeia original. Esses erros de alinhamento podem ser corrigidos pelo sistema de reparo, mas, se o erro não for reparado será gerado, no primeiro caso, um alelo com mais repetições e, no segundo caso um alelo com menor número de cópias.
Métodos de estudo dos microssatélites
A técnica fundamental de estudo de microssatélites é a PCR em combinação com técnicas adicionais como:
PCR + eletroforese: utilizam-se iniciadores que flanqueiam a região polimórfica. Após a amplificação, o produto de PCR é visualizado através de um gel de agarose e os distintos alelos são diferenciados pelo tamanho das bandas.
PCR + sequenciamento: um dos iniciadores da PCR é marcado com fluorescência no extremo 5 ́. Após a amplificação, os produtos marcados fluorescentemente são sequenciados em sequenciador capilar (o mesmo utilizado no sequenciamento de Sanger) permitindo a determinação do comprimento de cada fragmento. A marcação de distintos microssatélites com diferentes cores permite a sua genotipagem simultânea (técnica denominada multiplex).
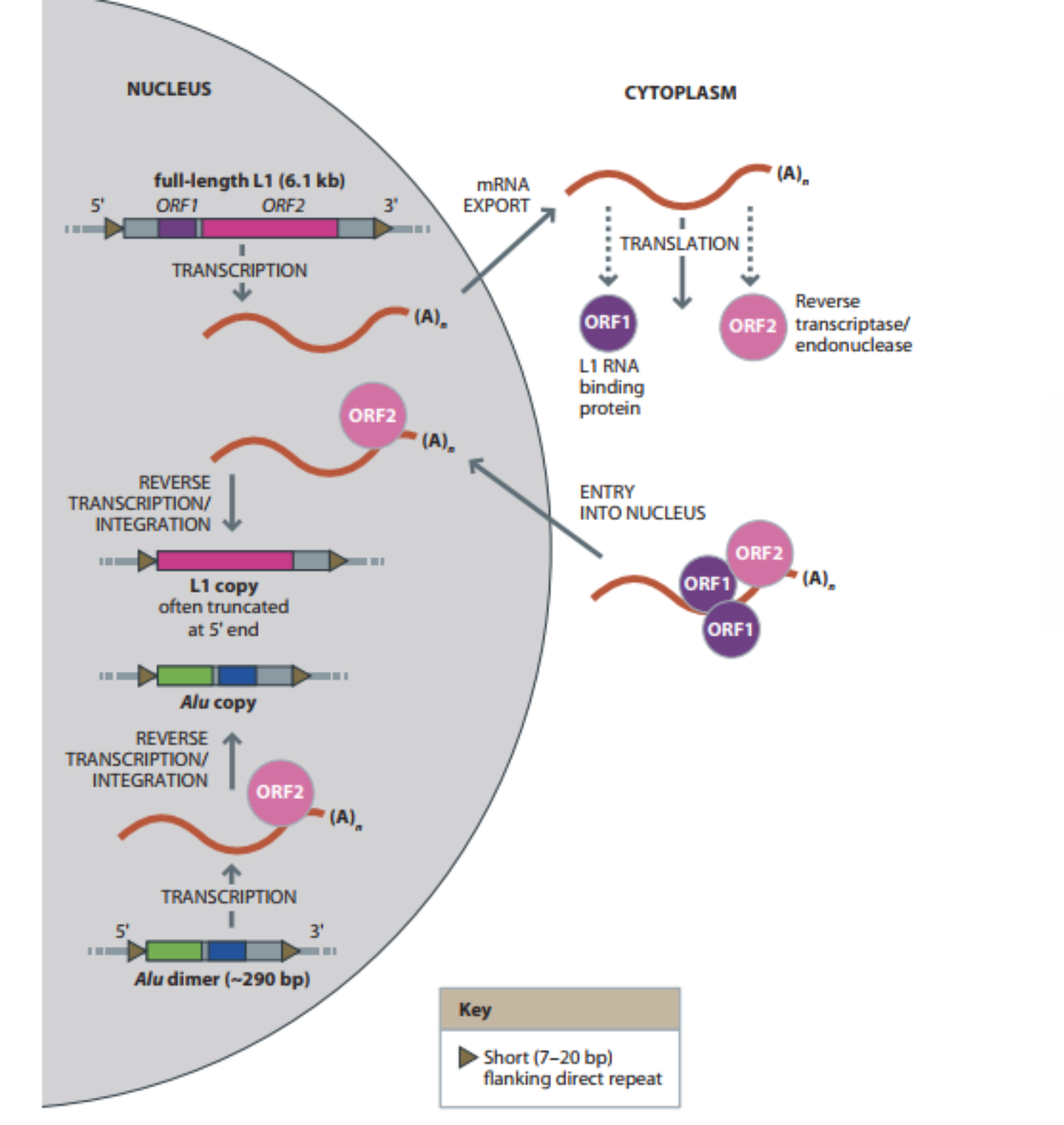
Inserções Alu
As inserções da família Alu são os elementos repetitivos dispersos mais bem estudados.
São retrotransposons curtos (~300pb) derivados de uma sequência original que se transpõe periodicamente a outras regiões do genoma. Transposições Alu têm ocorrido desde a origem dos primatas e por isso todos os humanos possuem certas cópias em regiões específicas do genoma (Alu não polimórficas ou fixas), enquanto outras ocorreram mais recentemente originando polimorfismo de inserção Alu (AIP). Nos humanos cerca de 10% do genoma é formado por esses elementos. A sua velocidade de transposição é muito baixa (ainda menor que a de SNPs) motivo pelo qual apresentam identidade por descendência. O estado ancestral é sempre a ausência da sequência Alu. Ocorrem nos autossomos e nos cromossomos X e Y. Existem >1 milhão Alus no genoma humano, mas apenas ~2000 são ativas.
Mecanismo de origem
As sequências Alu não codificam genes. Elas possuem sítios de reconhecimesão transcritas pela RNA polimerase III, quem realiza a sua transcrição. Para sua transposição elas dependem da maquinaria enzimática de um outro elemento transponível (L1). A endonuclease de L1 reconhece e corta uma sequência consenso gerando o sítio de inserção, enquanto a retrotrascriptase de L1 retrotranscreve o RNA Alu para DNA, que pode então ser inserido na nova posição.
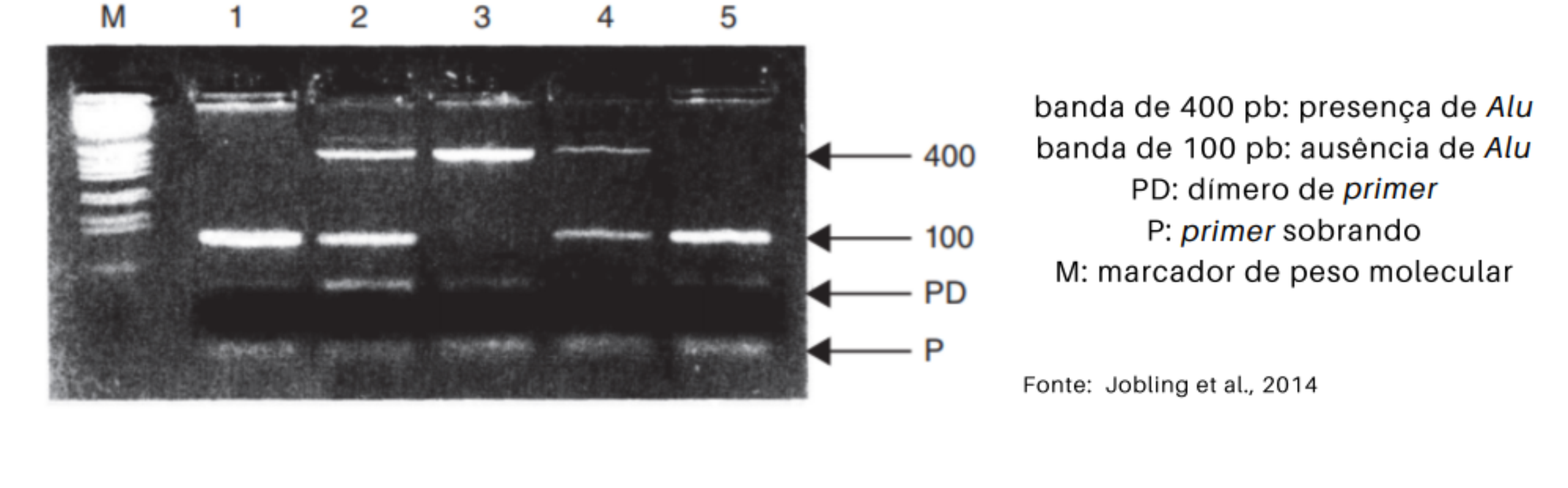
Método de estudo
Devido à presença de regiões consenso que flanqueiam o elemento Alu e a sua sequência relativamente curta, eles podem ser facilmente detectados por PCR + eletroforese em gel de agarose. Em presença da inserção, a PCR amplifica uma banda 300pb maior do que na ausência da mesma.
3. Sistemas uni e biparentais
Uma questão importantíssima que determina a aplicabilidade dos distintos polimorfismos, na prática, é a sua forma de herança. A mesma depende do cromossomo em que aquele polimorfismo está inserido.
Podemos dividir em:
Marcadores biparentais: autossomos, cromossomo X.
Marcadores uniparentais: cromossomo Y, DNAmt.
Marcadores biparentais
- Autossomos: 99,9% do DNA da célula.
- São os marcadores mais estudados para entender as bases genéticas de fenótipos.
- O seu estudo mostra a história completa de miscigenação de um indivíduo/população.
- Sofrem recombinação.
Marcadores Uniparentais
- DNAmt, cromossomo Y.
- Não sofrem recombinação, a única fonte de diversidade é a mutação.
- Uteis para conhecer linhagens.
- O seu estudo mostra apenas a história parcial de miscigenação de um indivíduo/população.
- Permitem detectar vieses sexuais nas contribuições ancestrais de uma população e outros fenômenos demográficos Ex.: patri ou matrilocalidade (em antropologia, descreve o comportamento das populações, se os casais tendem a morar na população de origem do individuo masculino ou femenino, respectivamente).
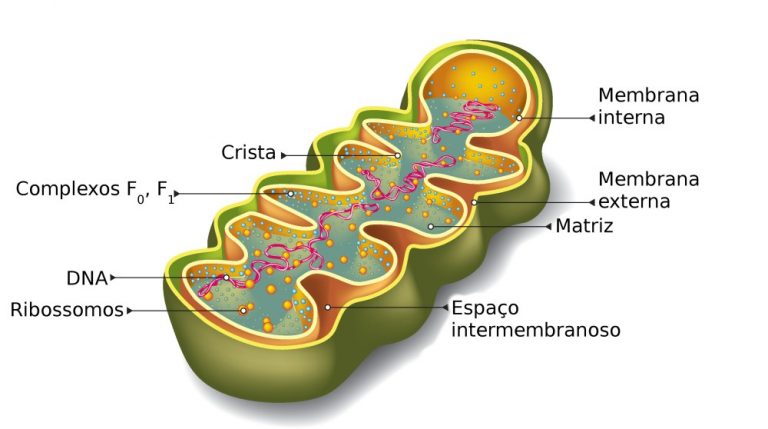
O DNAmt
O genoma mitocondrial tem sido amplamente utilizado no estudo de populações humanas devido a suas características:
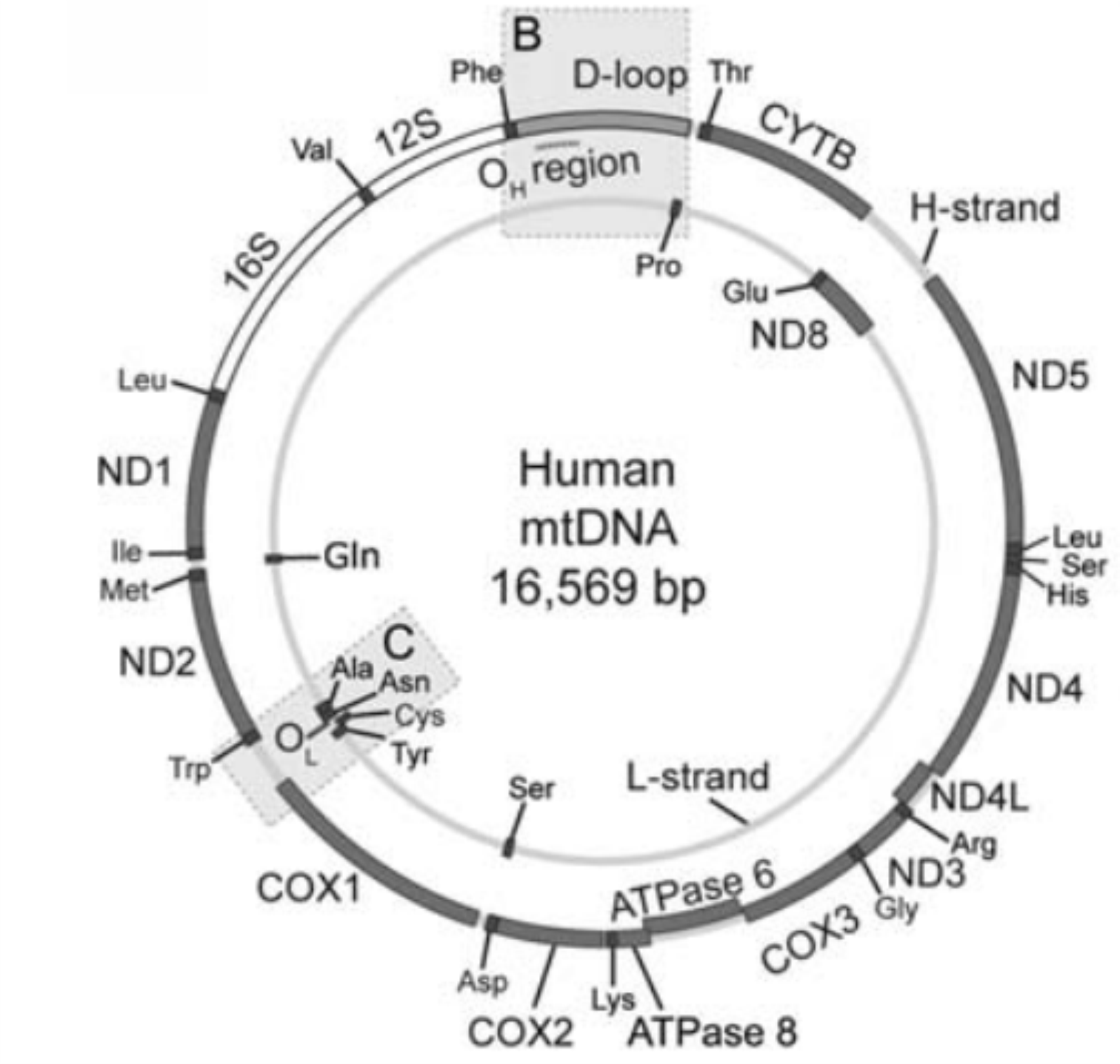
- Ele é um genoma haploide pequeno, 16.569 pb.
- Genoma circular, compacto e sem íntrons (DNA não codificador é apenas 7%).
- Não possui histonas.
- Codifica apenas 37 genes:
- 13 envolvidos na fosforilação oxidativa.
- 2 RNAr
- 22 RNAt.
- Código genético diferente do nuclear.
- Alto número de cópias (cada célula somática possui centos de milhares de cópias).
- Herança materna.
- Marcadores estudados: SNPs e indels, sequenciamento completo e/ou apenas da região de controle. Essa região contem os promotores de transcrição para os RNAm policistrônicos.
- Velocidade de mutação é 10 X maior do que a do genoma nuclear. É possível estimar essa velocidade diretamente desde pedigrís.
- Velocidade de mutação varia entre as distintas partes do DNAmt, sendo 10 X maior nas regiões hipervariáveis I e II (HVSI e HVSII) na região de controle. Isto devido a:
- Envolvimento da mitocôndria na fosforilação oxidativa que produz radicais livres mutagênicos.
- Maior taxa de replicação que o DNA nuclear.
- Em cada célula há uma coexistência de cópias wild type (de tipo selvagem) e mutadas = heteroplasmia (o oposto é homoplasmia). Porém, o indivíduo apresenta um genoma mitocondrial predominante (o mais comum).
O cromossomo Y
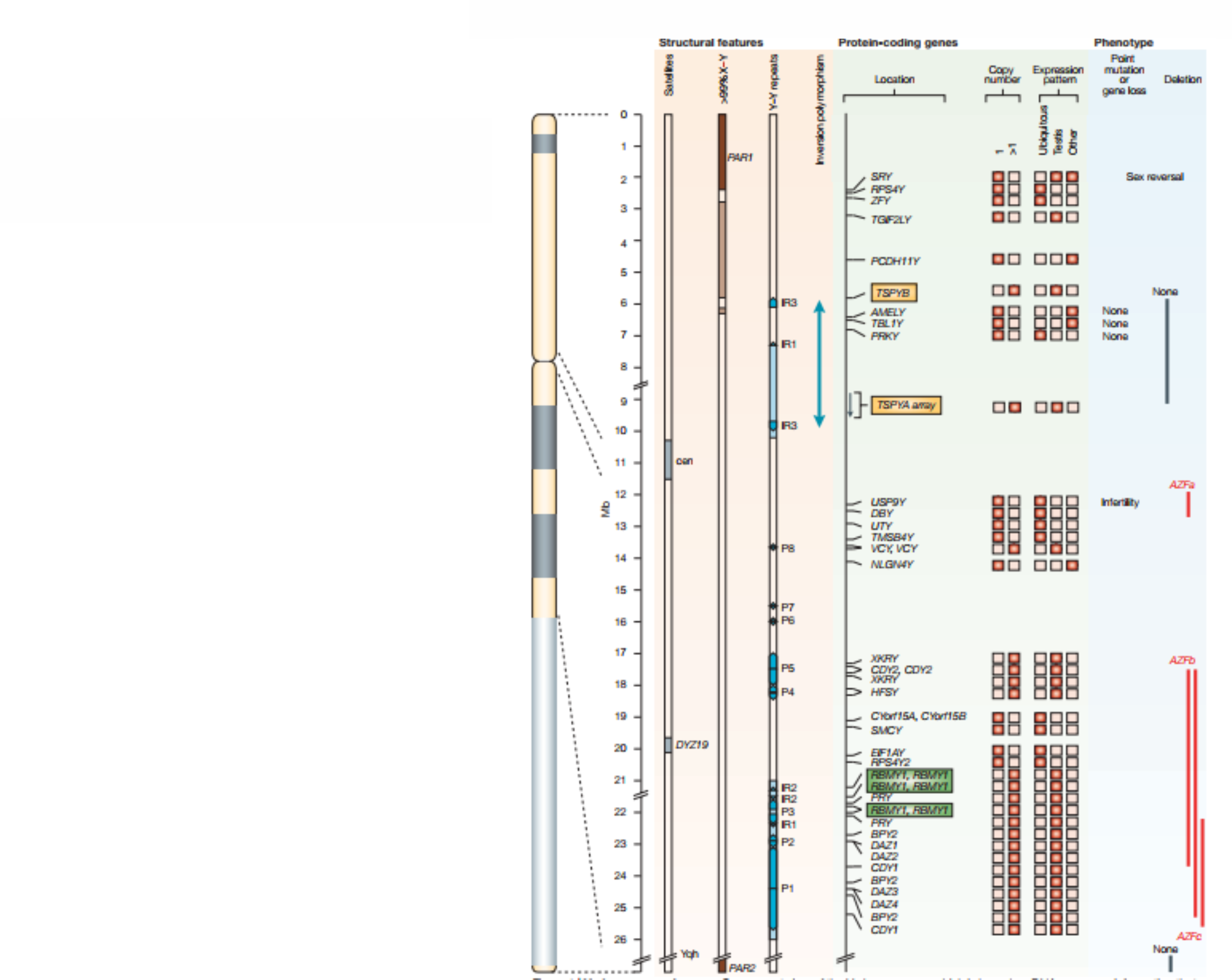
- Genoma haploide de 60 Mb (2% do genoma humano haploide).
- ~95 % DNA não recombinante (NRY = non-recombining Y), haplótipos são passados intactos de pai para filho.
- Única fonte de variação é a mutação.
- ~5% é constituído pelas regiões pseudoautossômicas (PAR1 e PAR2, teloméricas) as quais recombinam com regiões homólogas no cromossomo X.
- > 50% de DNA não codificante.
- Tendência a degenerar: ao longo da sua evolução foi perdendo genes.
- ~200 genes, ~55 ativos principalmente envolvidos com desenvolvimento sexual e fertilidade masculina. Alguns genes possuem homólogos no cromossomo X.
- Inclui o gene SRY, determinante do sexo.
- Marcadores estudados: STRs, Alu, indels, SNPs, sequenciamento completo.
4. Aplicações na antropologia
Marcadores biparentais
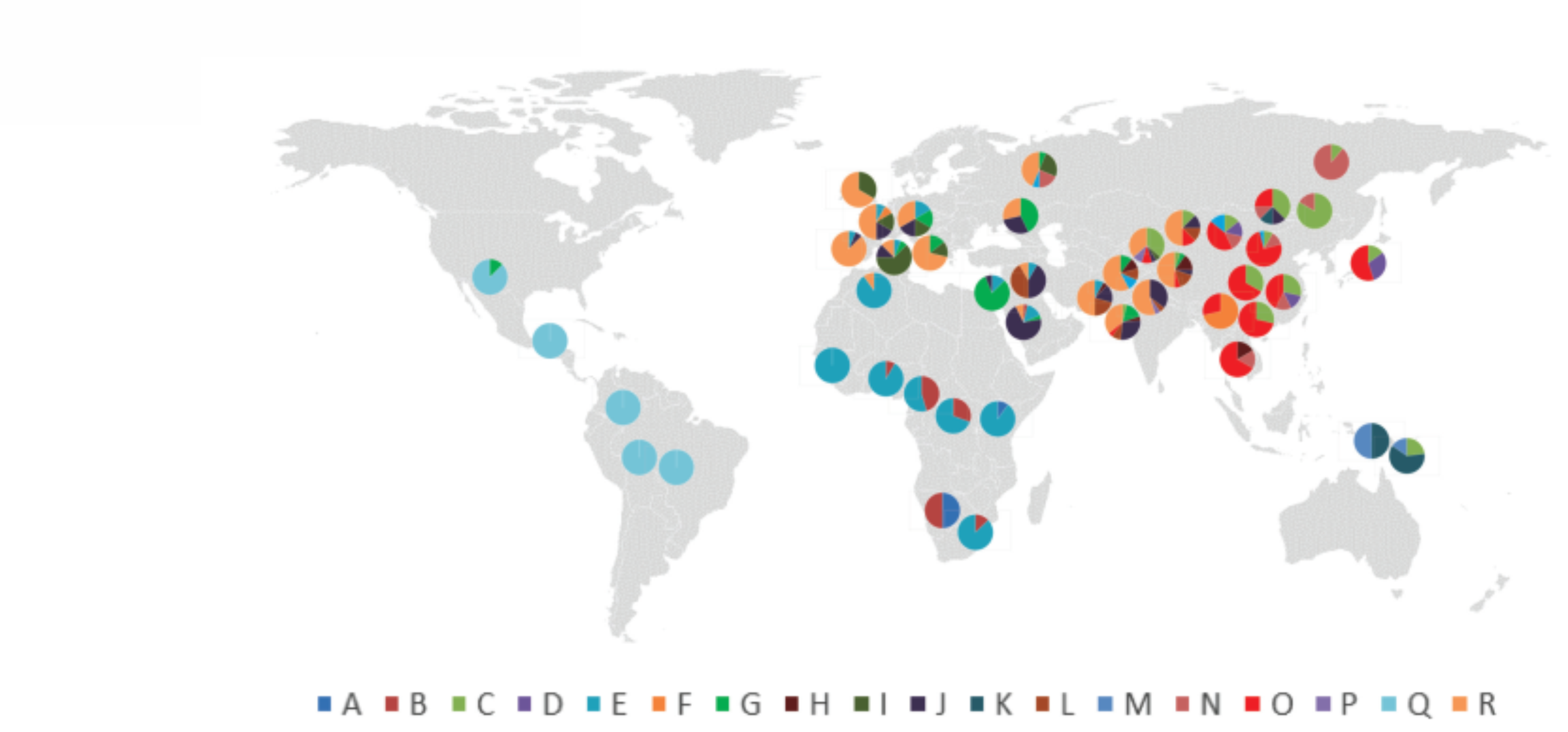
Em antropologia os marcadores biparentais podem ser utilizados para estudar a ancestralidade e estrutura populacional.
Marcadores utilizados:- Quando apenas pode ser genotipado um conjunto limitado de marcadores utilizam-se AIMs (ancestry informative markers) ou marcadores informativos de ancestralidade, os quais devem possuir diferenças altas de frequências entre as populações que se deseja investigar.
- Marcadores genômicos (Genome-Wide SNPs).
- O mais frequente é utilizar marcadores de velocidade de mutação baixa (Ex.: SNPs, Alu, indels) para estudos de ancestralidade e marcadores de velocidade de mutação alta (Ex.: microssatélites) para avaliar sub-estrutura populacional. Quando maior a resolução que se quer obter (por exemplo, ancestralidade no nivel continental e subcontinental) maior o número de marcadores necessários.
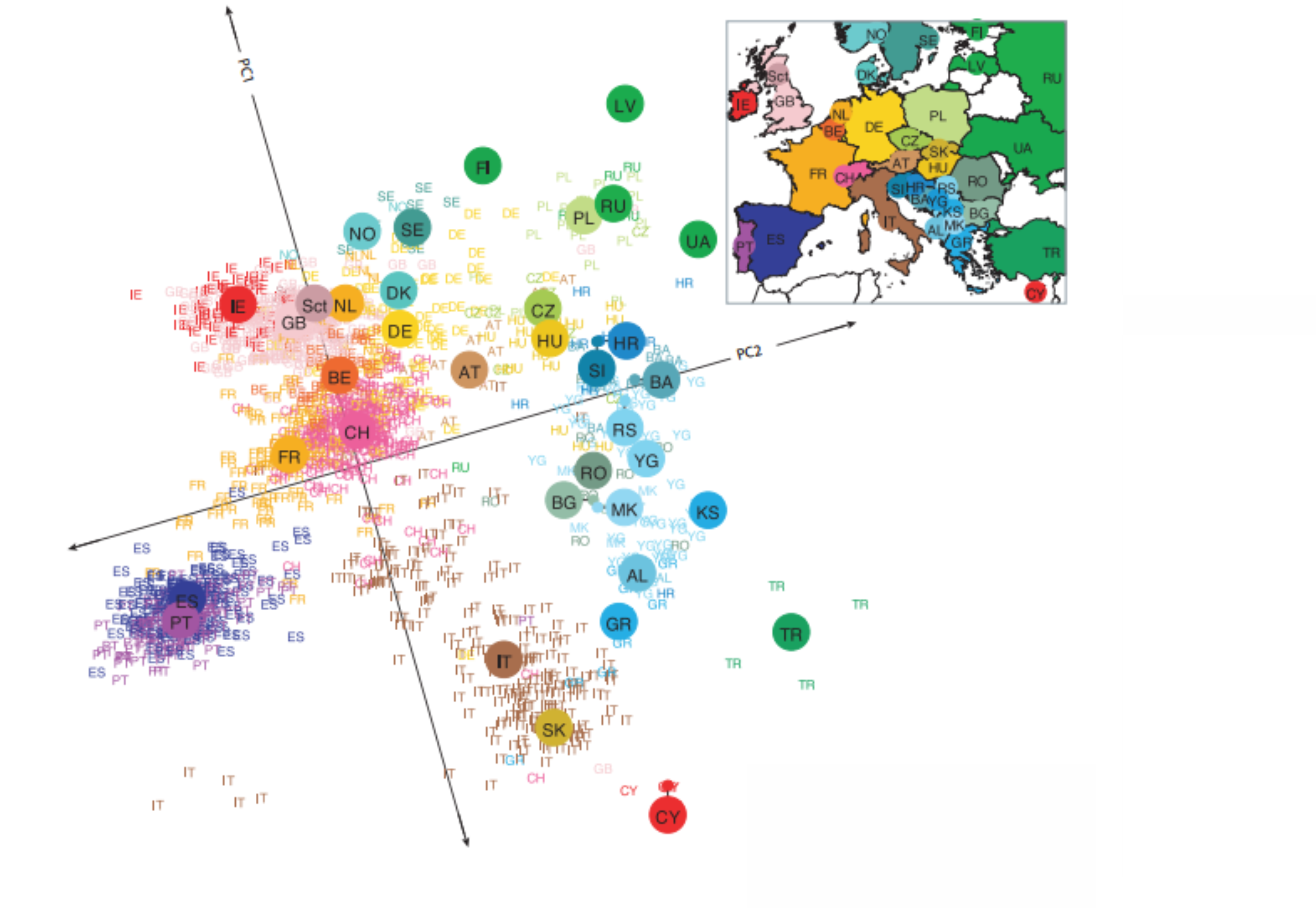
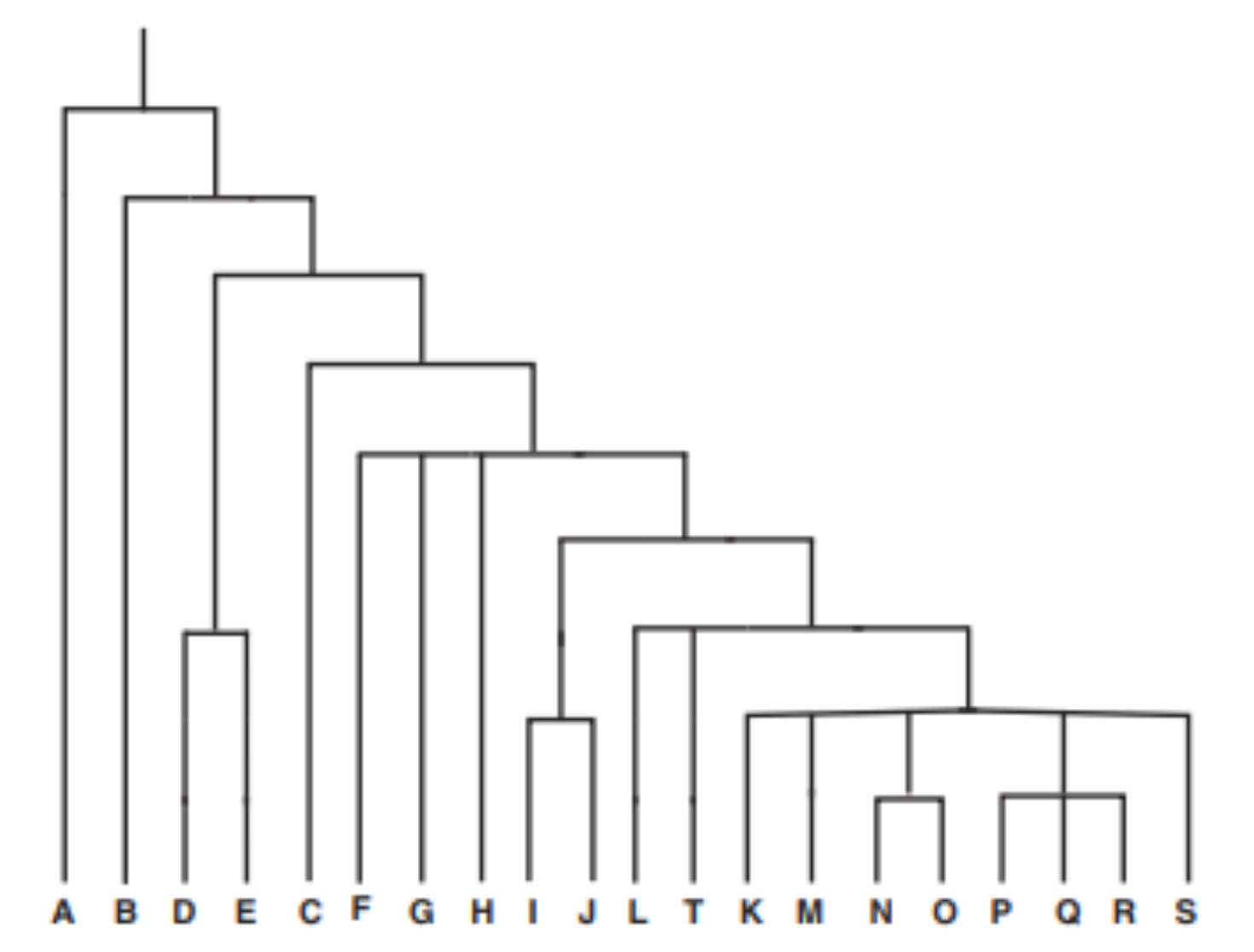
O DNAmt, devido às suas características, possui uma longa história de estudos na genética de populações. Ele foi sequenciado em 1981 (muitas décadas antes que o genoma nuclear, do ano 2000).
Cromossomo Y O seu uso é similar ao do DNAmt, porém mais limitado pela maior dificuldade de recuperação (menor número de cópia e maior suscetibilidade a degradação).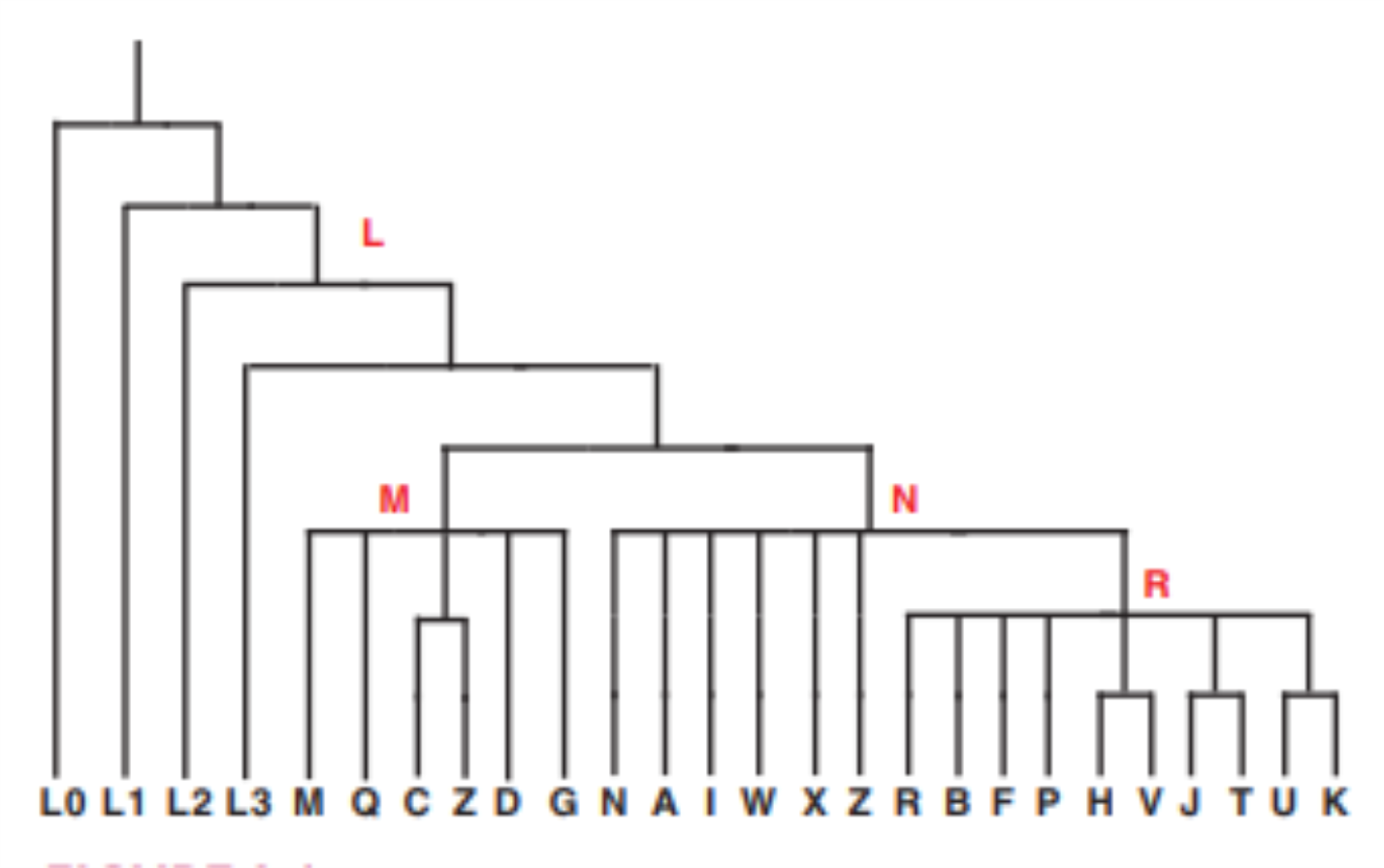
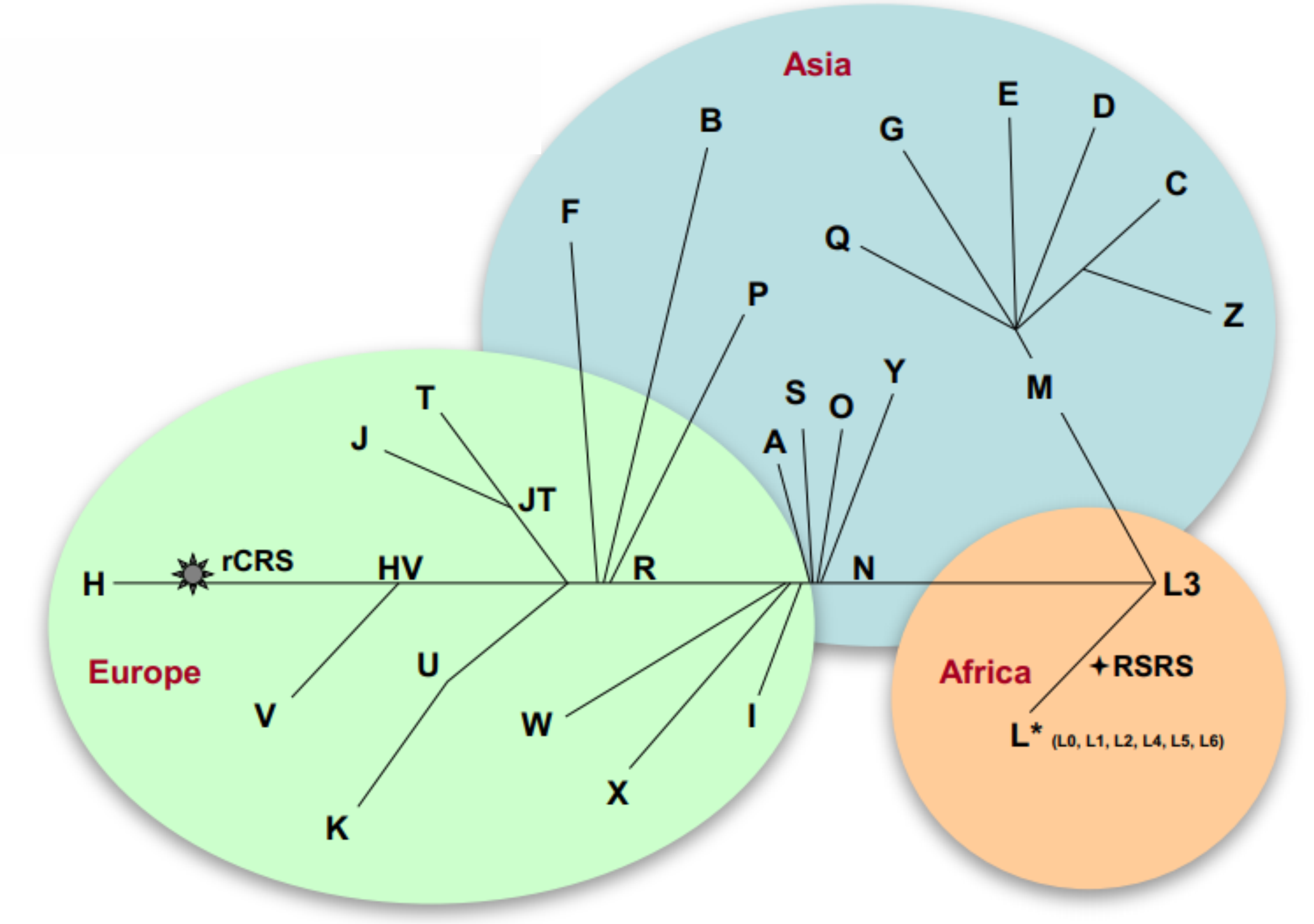
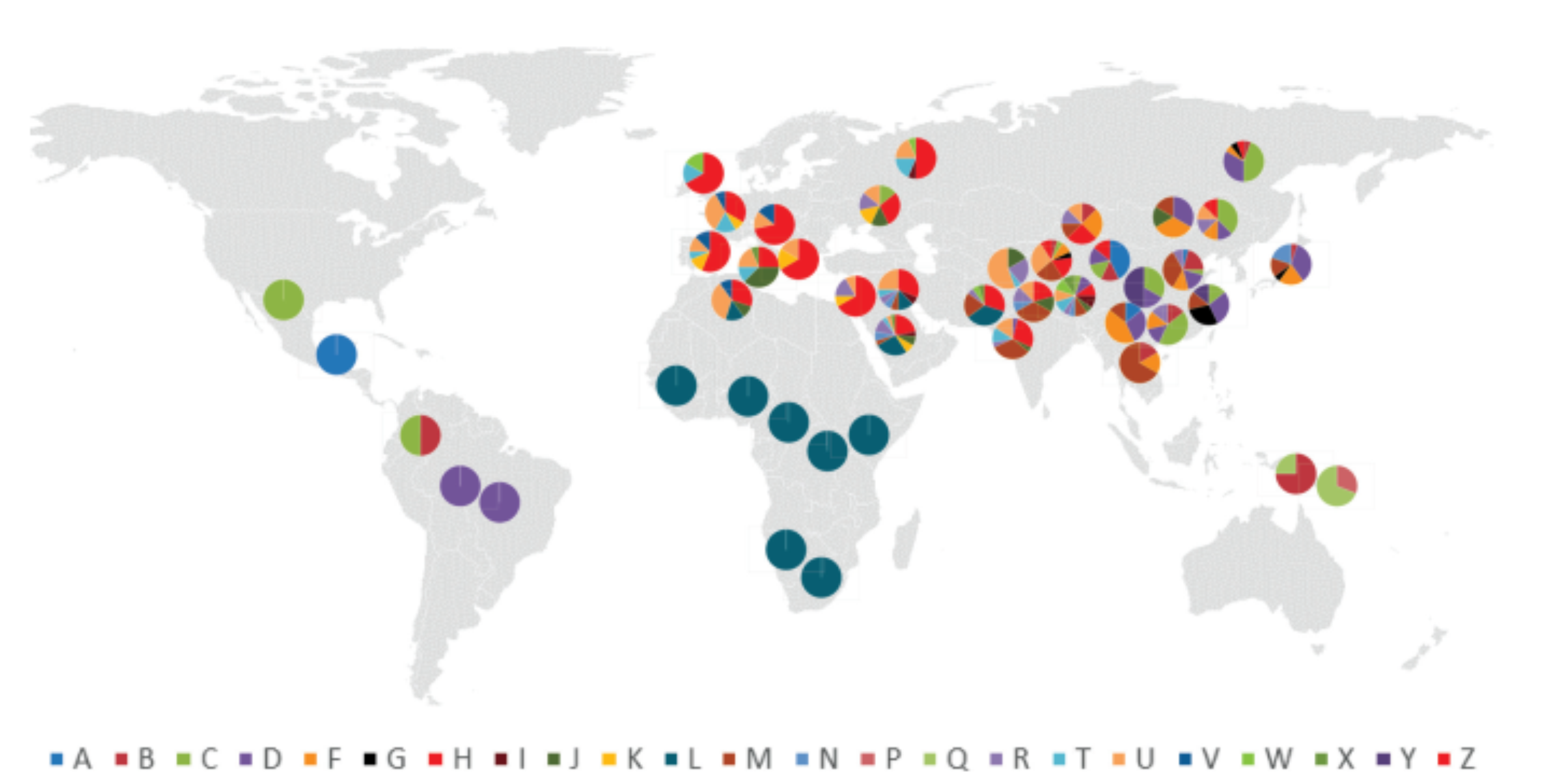

5. Aplicações na genética médica
Marcadores biparentais
Os marcadores autossômicos são os mais utilizados para estudos biomédicos devido a que eles são responsáveis pela codificação da maior parte das proteínas do organismo. Em função do conhecimento prévio que o pesquisador possui sobre o fenótipo de interesse podem ser feitos vários tipos de estudo para determinar as bases genéticas dos mesmos:
Marcadores uniparentais DNAmt O DNAmt é estudado quando existe alguma relação entre o metabolismo energético é o desenvolvimento da doença. É importante lembrar que graças à heteroplasmia, mutações patogênicas podem estar presentes em níveis baixos sem afetar a saúde (nível de mutação <1%). O fenótipo chega a ser evidente com o aumento da frequência da mutação patogênica, que pode produzir disfunção mitocondrial afetando principalmente tecidos com alto requerimento de ATP como músculo esquelético e cérebro. A severidade de doenças de origem mitocondrial depende do nível de heteroplasmia. Existem estudos de associação de haplogrupos com doenças, possivelmente pela sua relação com a ancestralidade. Cromossomo Y Os estudos biomédicos do cromossomo Y tem associado marcadores principalmente a doenças envolvendo fertilidade, hipertensão, câncer de próstata e testicular.
6. Aplicações nas ciências forenses
Marcadores biparentais
Nas ciências forenses os marcadores biparentais são especialmente utilizados para a identificação humana.
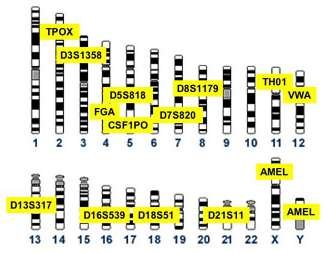
- Utilizados para identificação de suspeitos de ambos os sexos. É analisado um conjunto de STRs autossômicos distribuídos de forma a representar o genoma do indivíduo.
- Atualmente existem kits otimizados para uso forense.
DNAmt
- Especialmente útil na identificação a partir de espécimes forenses com DNA degradado (ossos, cabelo, dente).
- Mais estável por ser circular e estar mais protegido da degradação.
- Não serve para identificação de pessoas já que ele é compartilhado por todos os parentes por linha materna. Porém, ele pode ser usado na confirmação da identidade da pessoa.
- Utilizado na identificação de desaparecidos e investigação de desastres em massa.
- Devido ao seu alto número de cópias, a manipulação do DNAmt tem um alto de risco de contaminação pelo próprio pesquisador, devendo ser tomados cuidados extremos.

Cromossomo Y
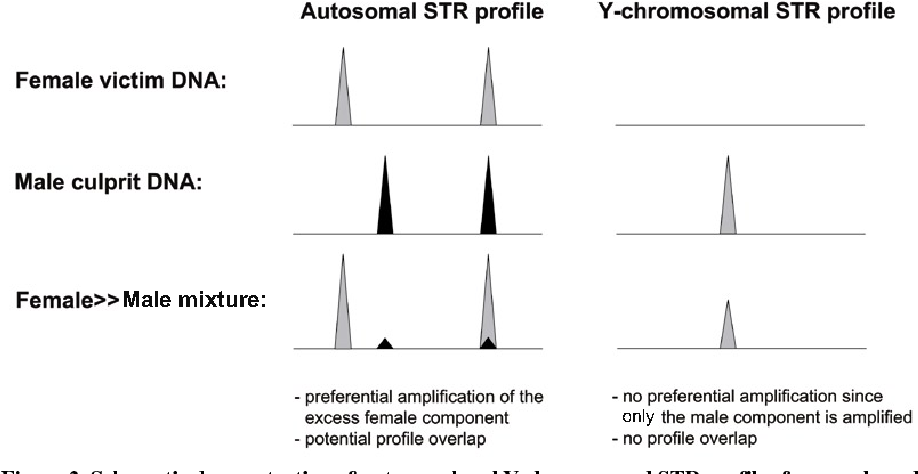
- Contribui na identificação de suspeitos masculinos através de um perfil de STR (Ex.: em casos de estupros).
- Não serve para identificação de pessoas, mas permite a exclusão de suspeitos.
- Compartilhado pelos parentes por linha paterna.
- Utilizado na sexagem de amostras pela amplificação de genes específicos do Y (Ex.: amelogenina) ou indels.
- Estimação de paternidade.